
While SaMD’s innovation has the potential to break new clinical ground, its newness causes headaches for regulators who do not know how to manage it. Any software used in a formal medical setting, meaning that its purpose is for a medical professional to treat, diagnose, or cure a patient instead of tracking their data, must be regulated. The FDA and other regulatory bodies are still working out how to approve software, particularly machine learning, but they have a few existing protocols in place.
The Regulators
The principal regulator for this type of protocol is the International Medical Device Regulators Forum, or IMDRF, a voluntary association of international medical regulators. The FDA, or Food & Drug Administration, plays a prominent role in the IMDRF and regulates SaMD usage in the United States. One crucial facet to appreciate is that SaMD products is still regulated under legacy medical device regulations, but is supported by guidelines (that do not meet the level of regulations) pursuant to the software in a medical device setting.
What Guidelines Exist?
The FDA and other governing bodies are still working through the guidelines for this new class of products. However, there are a few existing guidelines for clinical assessment of software, specifically when it is used in a clinical setting (consumer-based software falls under a different category).
Clinical guidelines include benefits and relevance. The manufacturer has to prove that the product provides a tangible benefit to patients, improves treatment outcomes, and is relevant to their medical goals. The device also has to be compatible with clinical language and terminology to integrate well with existing medical technology products.
| State of healthcare situation or condition | Significance of information provided by SaMD to healthcare decision | ||
| Treat or diagnose | Drive clinical management | Inform clinical management | |
| Critical | IV | III | II |
| Serious | III | II | I |
| Non-Serious | II | I | I |
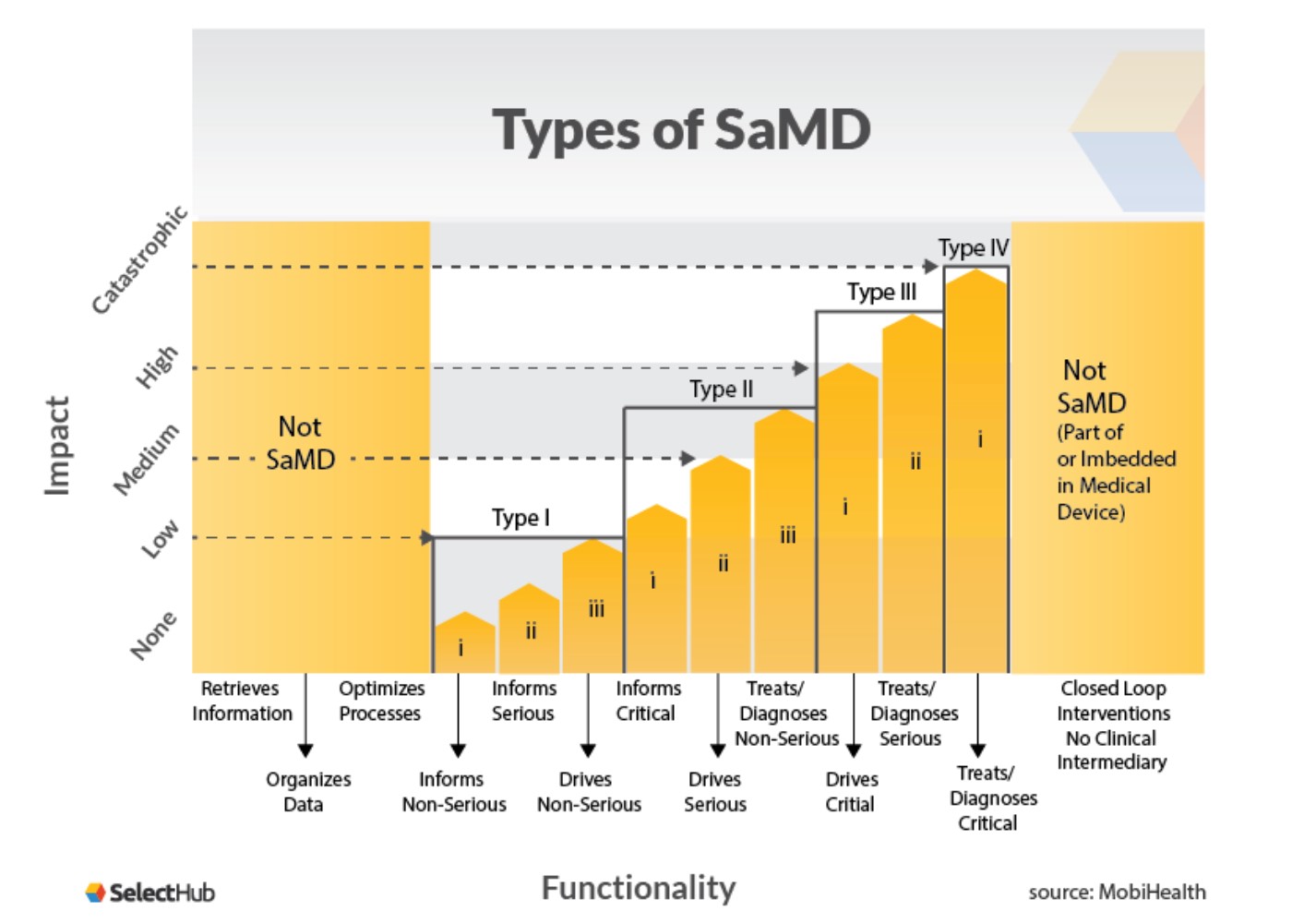
Once you have your definition statement, your SaMD can be determined to fit one of the categories I – IV. These are based on how vital the information provided by the SaMD is for patient or public health, with category IV being the highest level of impact.
For example, if the information provided by the SaMD treats or diagnoses critical healthcare conditions and is vital to avoid death or serious disability, then it will likely be categorized as an IV. If it simply informs a non-serious condition, then it is probably a category I. The diagram below from MobiHealth illustrates this:
What Does the Future of Software as a Medical Device Regulation Hold?
One of the biggest challenges for regulating Software as a Medical Device is that the software is constantly changing and developing even after approval, unlike traditional medical devices. This is true in a particular software that relies on artificial intelligence/machine learning.
In early 2021, the FDA published an action plan for approving AI/MI-based SaMDs. The action plan includes a “Predetermined Change Control Plan” designed for products evolving with usage. When manufacturers apply for initial approval, they will also submit anticipated modifications, implement those modifications to minimize risk for patients, and what monitoring the manufacturer will put in place during usage to protect patient privacy and data.
Guidelines, Not Regulations
So far, most of the regulation around SaMDs is not binding regulation but guidelines. Looking ahead, experts such as Crowell believe that the emphasis on policies instead of binding rules will continue. SaMDs are such a new field and not a priority in the FDA’s current regulation slate, so the government agency will likely leave more freedom to manufacturers.
Manufacturers are advised to focus on the FDA’s anticipated modifications protocol in the 2021 action plan, particularly real-time monitoring and addressing algorithmic bias. This will prepare a product to address the FDA approval mechanism.
The First: Caption Guidance
In February 2020, FDA issued its first authorization of an AI-backed SaMD, a cardiac ultrasound software. This was via the De Novo pathway and was approved in part due to the manufacturer’s use of a Predetermined Change Control Plan to incorporate future modifications.
When the FDA approved the product Caption Guidance back in 2020, it caused ripples in the software community because it was one of the first AI-powered software products to get formal approval. Caption Guidance is a software that enhances echocardiograms and helps users capture images of the heart and diagnose cardiac conditions.
One of the main benefits of Caption Guidance is that it makes diagnosis and image capturing accessible even to medical professionals that are not trained in this specific imaging technique. The software understands an acceptable image for diagnosis and guides health care professionals through the use of the machine.
Caption Guidance’s approval came through the above-mentioned De Novo pathway for uncategorized low-risk medical devices. Its example is essential not just for medical imaging software but also for other types of software.
Important Considerations
In addition to the usual concerns about functional software and regular updates, manufacturers of medical software need to keep other considerations in mind. One such existing regulation is HIPAA, which protects patient privacy, and manufacturers need to ensure that patient data will be anonymized and protected.
Another concern, including one brought up by the FDA, is algorithmic bias affecting machine learning outcomes. Algorithms rely on historical datasets that replicate existing biases against race, gender, class, and other factors affecting the quality of care—for example, existing algorithmic tools in medicine, such as risk-prediction algorithms, disadvantage Black people. Manufacturers of software must not replicate those biases in their software.
A Way Forward for Software as a Medical Device
Regulation pathways for SaMDs are not established yet as these programs are still new in the medical sphere. However, there is some precedent, such as the approval of Caption Guidance and many others. Manufacturers can apply for FDA regulation through the De Novo process.
For guidelines on conditions necessary for approval, manufacturers should look at the latest guidance from the IMDRF, as well as the FDA’s 2021 report. AI/MI-driven software must show proposed future changes, plan for implementing those changes without disrupting patients, and account for other factors such as algorithmic bias.
If these topics or others are of interest to you, we can help. Please call (650) 937-9164, or contact us here.
Recent Comments